Спинальная мышечная атрофия (СМА, проксимальная спинальная амиотрофия) – заболевание наследственной этиологии, которое характеризуется нарушением функционирования нервных клеток спинного мозга. Патология протекает с прогрессирующей слабостью мышц, атрофическими процессами в тканях, что в итоге вызывает полное обездвиживание. Заболеванию присвоен код по МКБ-10 G12.0 «Детская спинальная мышечная атрофия».
Причина патологии
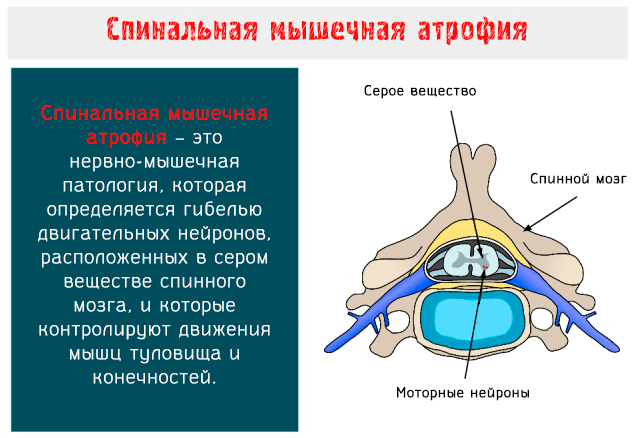 Основная причина возникновения спинальной мышечной атрофии – наследование дефектного гена аутосомно-рецессивным путем. Главное условие для его передачи – присутствие у обоих родителей. Диагностируют патологию 25% детей.
Основная причина возникновения спинальной мышечной атрофии – наследование дефектного гена аутосомно-рецессивным путем. Главное условие для его передачи – присутствие у обоих родителей. Диагностируют патологию 25% детей.
Возникновение болезни связано с дефицитом или полным отсутствием белка SMN, который участвует в выживаемости двигательных нейронов. Среди факторов риска:
- рождение мертвых детей у ближайших родственников по женской линии в анамнезе;
- смерть новорожденных детей в первый месяц жизни у ближайших родственников;
- наличие дефектного гена у близких родственников;
- диагностируемое заболевание у старших детей.
Спинально-мышечная атрофия у грудных детей встречается достаточно редко. Патологию диагностируют у одного младенца из 6-10 тыс. новорожденных. Заболевание мало изучено, плохо поддается диагностике и лечению. По данным статистики, примерно 30-35% таких малышей проживают не более 5 лет.
Первые сведения о спинальной мышечной атрофии появились в конце 19 века в творениях Вердинга-Гоффмана. Им были обнаружены патологические изменения в мышечных волокнах, периферических нервах, спинном мозге у малышей, которые родились с расстройством двигательной функции. Он отметил, что амиотрофия симметрична и развивается в передних рогах и передних корешках.
Клиническая картина
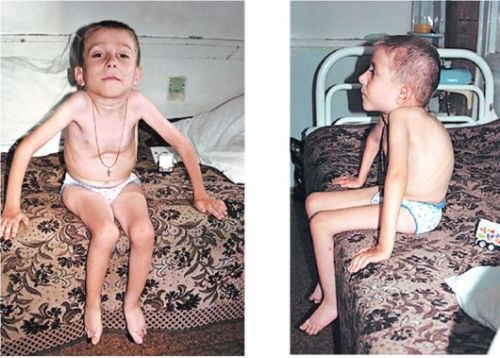
Клинические проявления патологии зависят от возраста, в котором развилось заболевание, и степени тяжести амиотрофии.
Симптомы спинальной мышечной атрофии:
- отсутствие рефлексов в ногах и руках;
- задержка в развитии: позднее сидение, стояние, ходьба или вовсе невозможность осуществления подобных действий;
- занятие позы «лягушка» маленькими детьми: в сидячем положении бедра отведены в стороны, а колени согнуты;
- снижение тонуса дыхательных мышц, слабый кашель, ослабленный крик и плач у новорожденного;
- застойный процесс в органах дыхательной системы, задержка бронхиального секрета, которая вызывает дыхательную недостаточность;
- тремор языка;
- нарушение процесса сосания, глотания, кормления.
 Выделены 4 формы заболевания, каждая из которых имеет свои симптомы:
Выделены 4 формы заболевания, каждая из которых имеет свои симптомы:
- Болезнь Вердинга-Гоффмана (1 тип). Начинает развиваться уже во время пренатального пребывания, первые симптомы возникают в 5-6 месяцев после рождения. Характерные признаки – слабость мышц, снижение рефлексов, выраженное затруднение процесса сосания, дыхания, глотания. Продолжительность жизни – до 1 года в 95% случаев. Летальный исход обусловлен дыхательной недостаточностью.
- Промежуточная амиотрофия Дубовица (2 тип). При заболевании второго типа первые клинические признаки обнаруживают в 3-15 месяцев от рождения. Менее 25% грудничков могут сидеть, но нарушен процесс ходьбы и ползания. Для патологии характерны симптомы паралича, выпадение глубоких сухожильных рефлексов, нарушение глотания. Многие больные уже к 2-3 годам становятся инвалидами и вынуждены находиться в инвалидной коляске. Продолжительность жизни при спинальной мышечной атрофии 2 типа – до 5 лет.
- Спинальная амиотрофия Кугельберг-Веландера (3 тип). Первые симптомы возникают в возрасте 15 месяцев-19 лет и схожи с клинической картиной, присущей 1 типу заболевания. Отличие – медленная прогрессия и лучший прогноз.
- Спинальная амиотрофия 4 типа или взрослая форма. Слабость мышц прогрессирует медленно, в патологический процесс в основном вовлекаются проксимальные мышцы.
У взрослых спинальную амиотрофию разделяют на следующие виды:
- болезнь Кеннеди (амиотрофический боковой склероз);
- болезнь Арана-Дюшенна (хронический полиомиелит взрослых);
- болезнь Шарко-Мари-Тута (моторно-сенсорная нейропатия с поражением дистальных отделов конечностей).
Характерный для всех форм патологии признак – мышечная слабость, которая со временем переходит в атрофию мышечных волокон. Причина тому – нарушение проводимости нервов и отсутствие сигнала к сокращению мышц, который исходит из спинного мозга.
Диагностика заболевания
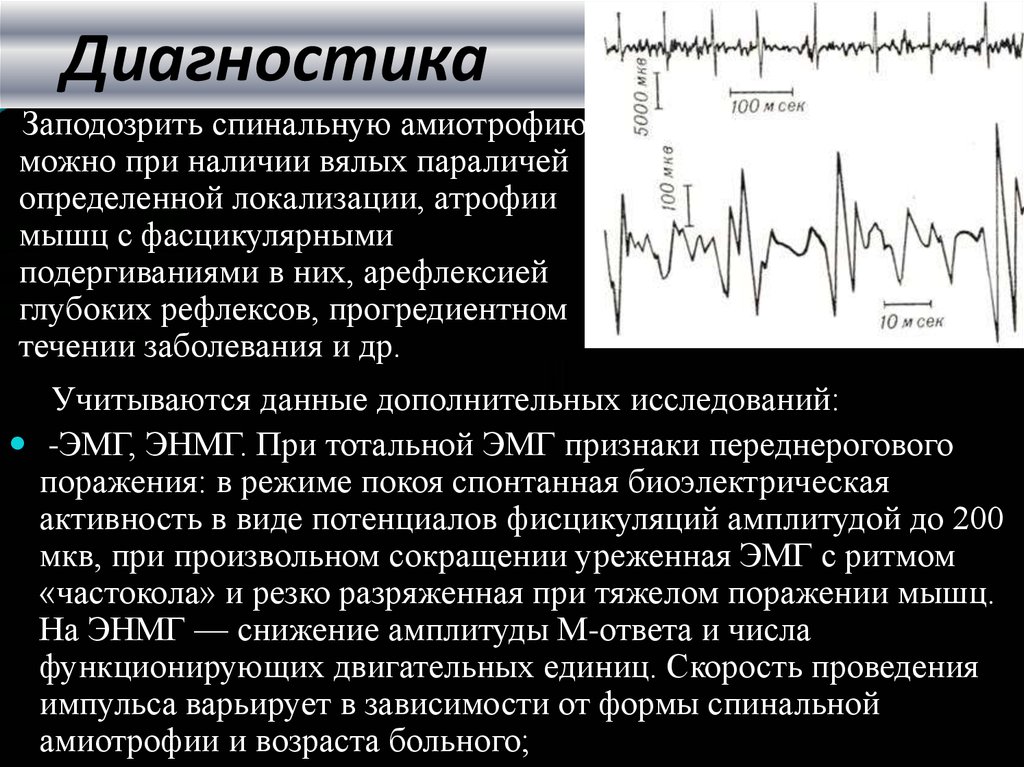 Клиническая картина спинальной атрофии не является решающим звеном в постановке диагноза. Подтверждают наличие заболевания по результатам генетического исследования. В 95% случаев патология развивается на фоне мутации – полной или частичной перестройки гена SMN. Для постановки диагноза достаточно выявления подобной мутации.
Клиническая картина спинальной атрофии не является решающим звеном в постановке диагноза. Подтверждают наличие заболевания по результатам генетического исследования. В 95% случаев патология развивается на фоне мутации – полной или частичной перестройки гена SMN. Для постановки диагноза достаточно выявления подобной мутации.
Генетический тест проводят с использованием биологического материала – крови. Может потребоваться проведение исследования крови на креатинфоскиназу, уровень фермента, который высвобождается из поврежденных мышечных структур. Такое тестирование не считается специфическим, так как фермент высвобождается и при других патологиях мышц.
Врач может назначить биопсию мышечных волокон, электромиографию, которая дает результаты об уровне биоэлектрических потенциалов, возникающих в мышцах.
Женщинам, которые имеют подобную генетику, рекомендовано проведение предимплантационной диагностики, используемой для скрининга пораженных эмбрионов (после экстракорпорального оплодотворения), и пренатальное тестирование (анализ ворсин хориона, бесклеточный анализ ДНК плода при беременности и др.).
Лечение заболевания
 Специальных лекарств и средств, которые помогли бы вылечить заболевание, до недавнего времени не существовало, поэтому его считали неизлечимым. Первый лекарственный препарат, которым сегодня можно лечиться от спинальной мышечной атрофии, зарегистрирован в России, имеет название «Спинраза» и способствует замедлению прогрессии патологии.
Специальных лекарств и средств, которые помогли бы вылечить заболевание, до недавнего времени не существовало, поэтому его считали неизлечимым. Первый лекарственный препарат, которым сегодня можно лечиться от спинальной мышечной атрофии, зарегистрирован в России, имеет название «Спинраза» и способствует замедлению прогрессии патологии.
Другие методы терапии позволяют восстановить неврологические функции:
- улучшить диапазон движений;
- увеличить мышечную силу;
- увеличить мышечную массу;
- улучшить баланс всего организма.
Хорошо зарекомендовало себя лечение стволовыми клетками. Биологический материал забирают из тканей пуповины и пуповинной крови. Биоптат получают от здоровых матерей после родоразрешения, подвергают его тщательной обработке перед введением реципиенту.
Получает донорский материал больной ребенок или взрослый внутривенно, методом инъекций через небольшую трубку. Так как стволовые клетки могут быть приняты системой кровообращения в каждом отделе туловища, они будут равномерно распределены по области поражения.
Практикуются интратекальные инъекции в область спинного мозга, его канала. Таким методом стволовые клетки быстрее и эффективнее проникают в спинной и головной мозг, в область поражения. Перед введением биологического материала проводят люмбальную пункцию для извлечения небольшого объема спинномозговой жидкости и замены таковой на биоптат.
Дополнительное лечение
 Полезно делать гимнастику, посещать массажиста, колоть или принимать внутрь специальные витамины. Последние необходимы для улучшения проводимости нервных окончаний. Это может быть тиамин, пиридоксин, аминокислоты. Также показан прием таблеток-ноотропов, сосудистых препаратов, средств, улучшающих проводимость мышц (альфа-липоевой кислоты, л-карнитина, Нейромидина и др.).
Полезно делать гимнастику, посещать массажиста, колоть или принимать внутрь специальные витамины. Последние необходимы для улучшения проводимости нервных окончаний. Это может быть тиамин, пиридоксин, аминокислоты. Также показан прием таблеток-ноотропов, сосудистых препаратов, средств, улучшающих проводимость мышц (альфа-липоевой кислоты, л-карнитина, Нейромидина и др.).
Рекомендуется специальная диета для детей, которые находятся на общем столе, и взрослых. В рацион должны входить растительные и животные белки. Полезно употребление каш, нежирного мяса, злаков, кисломолочной продукции, свежих фруктов и овощей. Пищу готовят на пару, в запеченном и отварном виде.
Полезные продукты в суточном меню распределяют так:
- фрукты и овощи – 50%;
- мясо и рыба – 35%;
- злаки и углеводы – 15;.
Из физиотерапевтических процедур эффективными будут сеансы электромышечной стимуляции, которые способствуют остановке дистрофических процессов. Существуют лечебные аппараты, предназначенные для использования дома. Особые усилия и специальные знания для самостоятельного применения не требуются.
Полезно проведение магнито- и лазеротерапии, электрофореза. Процедуры позволяют устранить болевой синдром при контрактурах, искривлении позвоночника и грудного отдела.
Прогноз и негативные последствия
Продолжительность жизни и прогноз были неутешительными до тех пор, пока на фармацевтический рынок не вышло инновационное лекарственное средство «Спиназа». Смерть многих новорожденных наступала сразу после появления на свет или на протяжении первого месяца жизни. При 1 типе заболевания нужна была постоянная респираторная поддержка. Максимальный срок жизни был около 1-2 лет.
Среди негативных последствий, которые часто диагностируются у детей и взрослых с диагнозом спинальная мышечная атрофия:
- бронхопневмония;
- аспирационная пневмония;
- острая дыхательная недостаточность.
Не нужно отчаиваться, если малышу поставили такой диагноз. Многие дети проживают 10 и более лет. Однако для достижения максимальных терапевтических результатов важен постоянный уход и круглосуточное наблюдение за ребенком, соблюдение назначений и рекомендаций лечащего врача.